Dra. María Hernández Valladares
- Categoría Profesional Actual: Contratada de Investigación Postdoctoral (Maria Zambrano Sr.)
- Código ORCID: 0000-0001-9347-7841
- Correo Electrónico de contacto: @email
- Teléfono de contacto: +34958241000 ext. 20284
- Directorio UGR: Acceso Información institucional Directorio UGR
Breve Resumen de la Carrera Investigadora de la Dra. Hernández Valladares:
María del Carmen Hernandez Valladares es investigadora del Departamento de Química Física contratada con ayudas “María Zambrano” para la atracción de talento internacional en la modalidad senior. También es miembro del Instituto de Biotecnología, del ibs.GRANADA y de la Unidad de Excelencia de Química Aplicada a Biomedicina y Medioambiente de la Universidad de Granada.
Obtuvo el doctorado en Bioquímica por la Universidad de Lieja (Bélgica) en 1998 sobre el rol de los iones Zn(II) en la estabilidad y actividad de la metalo-beta-lactamasa de Aeromonas hydrophila AE036. Entre 1999 y 2004 realizó una estancia postdoctoral en el International Livestock Research Centre (ILRI) en Nairobi (Kenya) bajo la supervisión del Pr. Fuad Iraqi sobre las bases genéticas de resistencia a Plasmodium en modelos de ratón. A continuación, realizó otra estancia postdoctoral en el Institute of Molecular and Cell Biology en Singapur bajo la supervisión del Pr. Bob Robinson para la cristalización y determinación de estructuras 3D de complejos de proteínas de CapZ y sus inhibidores.
Se inició en el campo de la big omics con espectrometría de masas en el año 2009 gracias a una estancia postdoctoral en el Department of Physiology de la Universidad de Liverpool (UK) bajo la supervisión del Pr. Ian Prior caracterizando el phosphosignaling cascade de las oncoproteinas Ras. Desde el año 2014 formó parte de la Plataforma de Proteómica de la Universidad de Bergen (PROBE) dirigida por el Pr. Frode Berven, en donde trabajo como investigadora sobre prognosis de pacientes con leucemia mieloide aguda con el Pr. Øystein Bruserud entre otros numerosos proyectos y en la Plataforma de Metabolómica, de la cual fue directora, de la misma Universidad desde el año 2021. Durante los años 2018-2019 trabajo en la prestigiosa Plataforma de espectrometría de masas de la Universidad de Sydney en Australia, en colaboración con el Pr. Stuart Cordwell, para el aprendizaje de diversas técnicas de caracterización de modificaciones post-translacionales (PTMs), metabolómica y lipidómica.
Líneas de Investigación desarrolladas por el equipo de la Dra. Hernández Valladares
La Dra. Hernández Valladares se encuentra desarrollando diversas líneas de investigación, entre las que destacan:
Cambios de proteoma y fosfoproteoma asociados a la prognosis de leucemia mieloide aguda.
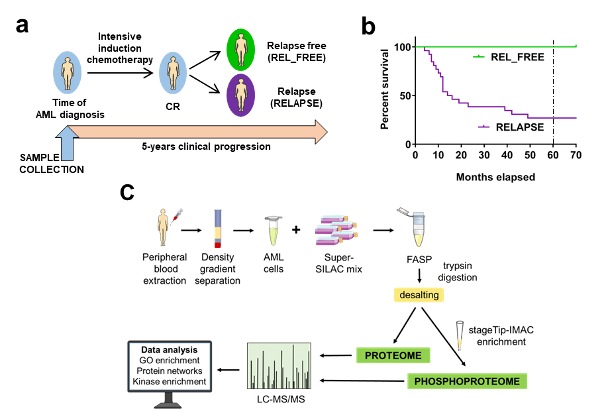
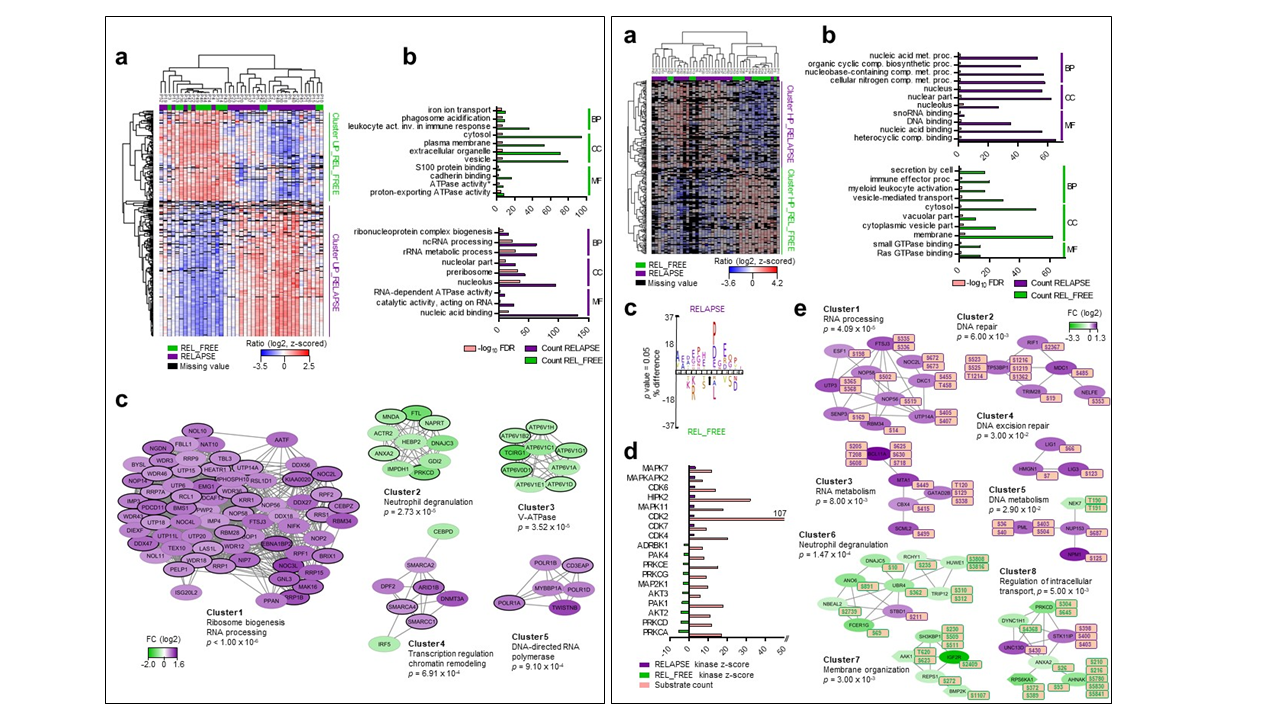
La leucemia mieloide aguda (LMA) es un cáncer hematológico que afecta principalmente a personas de edad avanzada. Aunque la remisión completa (RC) se logra para la mayoría de los pacientes después de las terapias de inducción y consolidación, casi dos tercios recaen en un intervalo corto. La comprensión de los factores biológicos que determinan la recaída se ha vuelto de gran interés clínico en la LMA. Utilizamos espectrometría de masas en tándem con cromatografía líquida (LC-MS/MS) para identificar los cambios de proteínas y los eventos de fosforilación de proteínas asociados con la recaída de LMA en células primarias de 41 pacientes con LMA en el momento del diagnóstico. Los pacientes se definieron como libres de recaídas si no habían recaído dentro de un seguimiento clínico de cinco años después del diagnóstico de LMA. La recaída se asoció con una mayor expresión de proteínas de procesamiento de ARN y una disminución de la expresión de proteínas V-ATPasas. También observamos un aumento en los eventos de fosforilación catalizados por quinasas dependientes de ciclina (CDK) y caseína quinasa 2 (CSK2). La relevancia biológica de los hallazgos del proteoma fue respaldada por ensayos de proliferación celular que utilizaron inhibidores de V-ATPasa (bafilomicina), CSK2 (CX-4945), CDK4/6 (abemaciclib) y CDK2/7/9 (SNS-032). Mientras que la bafilomicina inhibió preferentemente las células de los pacientes con recaída, los inhibidores de la quinasa fueron menos eficaces en estas células. Esto sugiere que la terapia contra las quinasas reguladas también podría dirigirse a los factores que inducen su regulación positiva en lugar de su actividad. Este estudio, por tanto, presenta marcadores que podrían ayudar a predecir la recaída de la LMA y orientar estrategias terapéuticas.
Estudios proteómicos y fosfoproteómicos en pacientes de leucemia mieloide aguda durante el tratamiento estabilizador de ácido retinoico todo-trans y ácido valproico
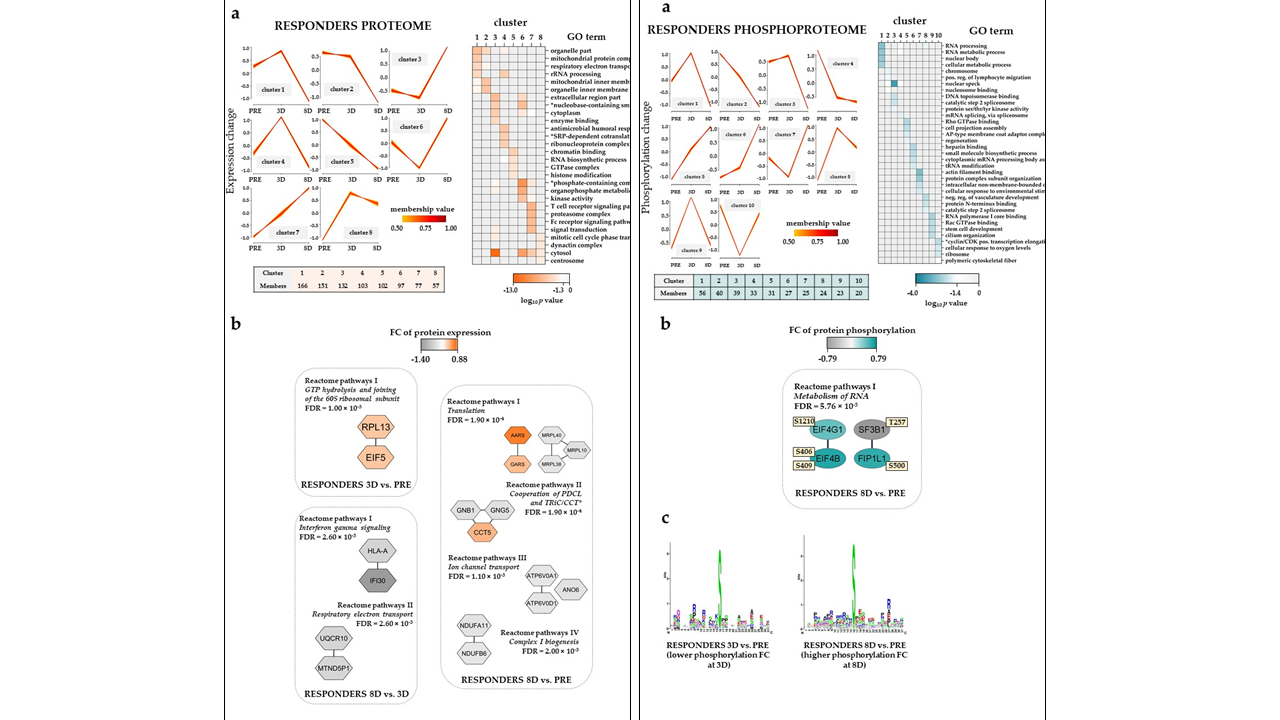
El ácido retinoico todo trans (ATRA) y el ácido valproico (VP) se han probado en el tratamiento de variantes no promielocíticas de la leucemia mieloide aguda (LMA). Los estudios no aleatorios sugieren que los dos medicamentos pueden estabilizar la LMA y mejorar los recuentos normales de células sanguíneas periféricas. En este contexto, utilizamos una estrategia proteómica/fosfoproteómica para investigar los efectos in vivo de ATRA/VP en células humanas de LMA. Antes de comenzar el tratamiento combinado, los pacientes con LMA que respondieron mostraron niveles elevados de varias proteínas, especialmente aquellas involucradas en la desgranulación/diferenciación de neutrófilos, la regulación de la fase M y la interconversión de nucleótidos, di- y trifosfatos (es decir, síntesis y unión de ADN). Varios entre los sitios de fosforilación regulados diferencialmente reflejaron diferencias en la regulación del metabolismo del ARN y eventos apoptóticos en el mismo punto de tiempo. Estos efectos fueron causados principalmente por el aumento de la actividad de la quinasa 1 y 2 dependiente de ciclina (CDK1/2), la quinasa de dominio LIM 1 y 2 (LIMK1/2), la proteína quinasa 7 activada por mitógeno (MAPK7) y la proteína quinasa C delta (PRKCD) en células de pacientes respondedores. Un efecto extenso del tratamiento in vivo con ATRA/VP fue el nivel alterado y la fosforilación de proteínas involucradas en la regulación de la transcripción/traducción/metabolismo del ARN, especialmente en los pacientes que no responden, pero la regulación del metabolismo celular, el sistema inmunológico y las funciones del citoesqueleto fueron también afectadas. Nuestro análisis de muestras seriadas durante la primera semana de tratamiento sugiere que los perfiles proteómicos y fosfoproteómicos pueden usarse para la identificación temprana de los pacientes respondedores al tratamiento basado en ATRA/VP.
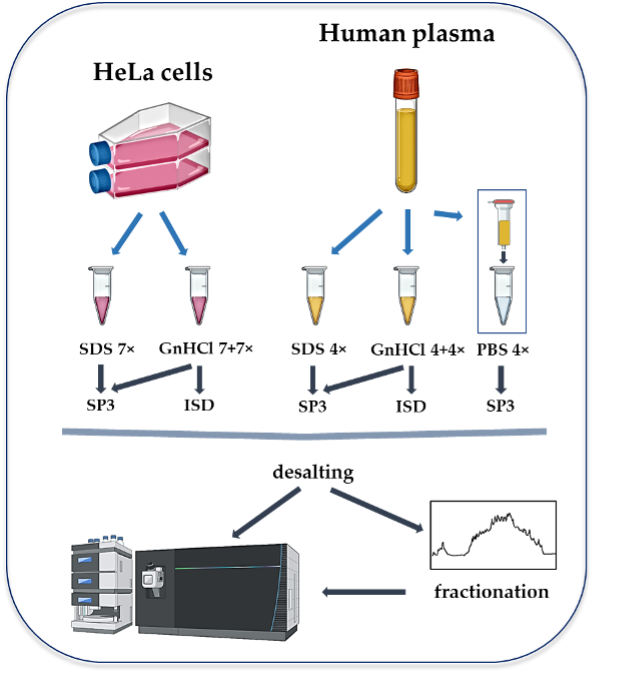
Optimización de workflows the LC-MS para el estudio proteómico de muestras de plasma y suero. Desarrollo de pipelines de biomarcadores en plasma y suero
El uso de una metodología adecuada de procesamiento de muestras para obtener la máxima cobertura de proteoma y datos cuantitativos de alta calidad es una elección importante antes de iniciar un estudio de proteómica basado en cromatografía líquida-espectrometría de masas (LC-MS). Los workflow de procesamiento de muestras para la proteómica implican la digestión del proteoma en solución o la preparación de muestras en fase sólida (SP3). Los probamos tanto en células HeLa como en muestras de plasma humano, utilizando tampones de lisis que contenían SDS o clorhidrato de guanidinio. También estudiamos el efecto del uso de minicolumnas de “depletion” comercialmente disponibles antes de SP3, para aumentar la cobertura del proteoma en muestras de plasma humano. Nuestros resultados muestran que el protocolo SP3, utilizando cualquiera de los dos tampones, logra el mayor número de proteínas cuantificadas tanto en las células HeLa (cerca de 8,000 proteínas) como en las muestras de plasma. Además, el uso de minicolumnas de “depletion” antes de SP3 da como resultado un aumento del doble de las proteínas plasmáticas cuantificadas. Con un fraccionamiento adicional, cuantificamos casi 1,400 proteínas y examinamos las proteínas de menor abundancia involucradas en las vías neurodegenerativas y el metabolismo mitocondrial. Por lo tanto, recomendamos el uso de la metodología SP3 para el procesamiento de muestras biológicas, incluidas aquellas posteriores al proceso de “depletion” de proteínas plasmáticas de gran abundancia.